Main Menu (Mobile)- Block
Main Menu - Block
homepage_slideshow | views
janelia_helper-janelia7_publication_list | block
Publications
01/24/23
|
01/24/24
|
janelia_helper-janelia7_view_all_publications | block
janelia7_blocks-janelia7_featured_stories_grid | block
Featured

SCIENCE NEWS
04/08/2024
|
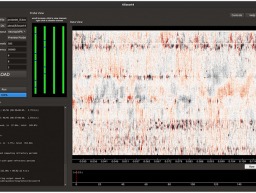
Janelia scientists release state-of-the-art spike-sorting software Kilosort4

INSTITUTE NEWS
03/28/2024
|
Renowned neurosurgeon Alfredo Quiñones-Hinojosa captivates Dialogues audience with stories of resilience


RESEARCH HIGHLIGHT
03/20/2024
|
In flies, a single brain cell can drive multiple movements of the body

RESEARCH HIGHLIGHT
02/21/2024
|
Researchers probe protein changes in the cell’s scavenger centers to understand longevity
janelia7_blocks-janelia7_featured_stories2_grid | block
custom | custom
janelia7_blocks-janelia7_homepage_promo | block